|

Merging Within-Chip Replicate Measurements
Overview
Certain import templates allow you to merge replicate genes occurring on the same chip into a single measurement. When this is done, GeneLinker™ uses the spread between the replicates to estimate a reliability measure for the resulting (average) measurement.
The statistical method used to merge replicate genes and generate a reliability measure is designed for use with small numbers of replicates (as few as two) and to give usable results even if there are missing data. To achieve this, the method assumes that the variability between the replicate measurements increases proportional to the abundance of the gene product, but otherwise has a roughly normal (Gaussian) distribution which is the same across all genes on the chip.
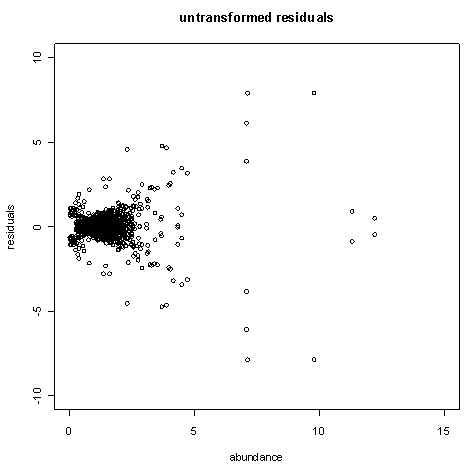
The figure below plots the difference between replicates against the average abundance (in arbitrary units) for a typical experiment with within-chip duplicate measurements. Notice that genes with greater abundance tend to have greater difference between the replicates.
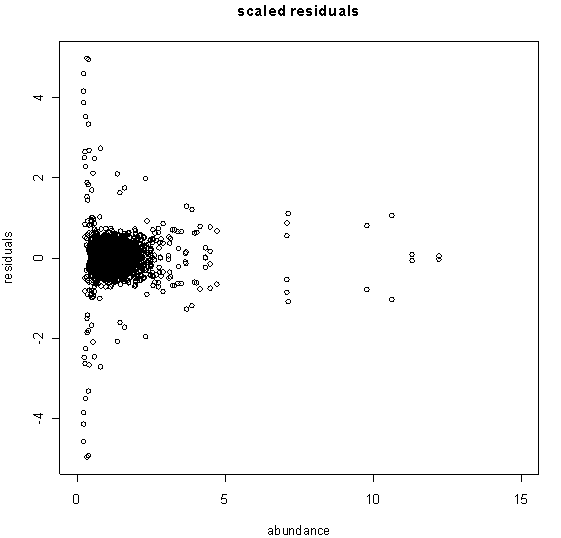
By scaling the replicates according to the abundance, we obtain the plot in the figure below. Note how the scaled residuals tend to be large when the average abundance is near zero. This is to be expected since measurements near the detection threshold are relatively more error-prone.
The resulting distribution of residuals has the shape of a 'bell curve' but has very long tails representing measurements with abnormally high variation between the replicates. In statistical terms, this example has a very large kurtosis.
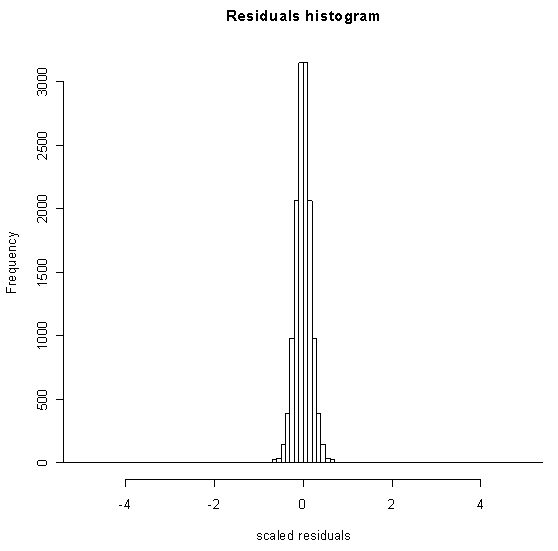
The integral of the tails of this distribution can be interpreted loosely as the probability of getting such an extreme residual by chance. We compute this probability and then take its complement in order to put this reliability measure on the same scale as the P-values many researchers are accustomed to. A value near zero means a reliable measurement; a value near one means an unreliable measurement.
Detailed Algorithm Used to Merge Within-Chip Replicate Measurements On Import
Here is a detailed description of the algorithm used to merge within-chip replicate measurements on import.
1. Read x[chip,gene,rep] from datafile
2. Compute abundance[chip,gene] = mean(x[chip,gene,:])
3. Save the abundance as the GeneLinker™ expression measurement
4. Compute resid[chip,gene,rep] = (x[chip,gene,rep]-abundance[chip,gene])/abundance[chip,gene].
These are the residuals plotted in the Figures 2 and 3 above.
5. Compute s = stdev(resid[:,:,:])
6. Set r[chip,gene] = max(abs(resid[chip,gene,:])) and compute the integral under the normal curve N(0,s) between -|r| and +|r|.
This step is quite conservative if you have more than three replicates, essentially taking the most extreme replicate as an indicator of the quality of the whole set.
7. Save this integral p[chip,gene] as the GeneLinker™ reliability measure
If due to missing data there are no replicates for a given chip/gene pair, then that measurement is arbitrarily assigned a reliability measure of zero (perfectly reliable). Therefore measurements for which you have no reliability information will not be filtered out by the Value Removal by Reliability Measure operation.
Naturally the assumptions of this model may be tested if you have enough replicates for each condition and gene. If you have more than three replicates and you feel this model is inappropriate, we recommend you use general-purpose statistical software to preprocess your data outside GeneLinker™, merging replicates before importing it in tabular format. You may eliminate unreliable measurements from the dataset before using the Tabular import template, or you may compute reliability measures and import them along with the expression data using the Tabular with Reliability Measures import template.
Related Topics:
Creating a Table View of Reliability Data
Removing Values by Reliability Measure